Next-Generation Sequencing
Once collected, sequencing the sample begins. The sample is removed from storage, and a portion is taken to prepare what is called a "library" for sequencing. This is always performed in a wet lab environment, and the chemical methods can be quite complex.
While it sounds easy to read the entire genome by laying all of the chromosomes end-to-end, in practice, there are some problems with this approach. Chiefly, researchers currently lack the biochemical processing abilities to extract and to read such long stretches of DNA quickly and accurately. Additionally, next generation sequencing technologies have their own set of limitations, including:
- Each nucleotide the sequencer calls is accompanied by a confidence score estimating how likely the recorded call is to be correct. In short, the sequencer's calls are of a probabilistic nature and do not have a zero error rate.
- As the sequencer observes each nucleotide along the read, the confidence it has in each new call drops so that if allowed to continue eventually the data quality of the last bases would be too low to use reliably.
- Sequencers can only read DNA fragments because RNA is too unstable. So biochemical methods are employed to reverse the transcription process and produce a DNA fragment that captures the information in the RNA sequence.
Given these physical limitations, this process, known as shotgun sequencing, looks like the following:
- The sample's DNA is fragmented into billions of smaller DNA pieces that are somewhere between tens to thousands of bases long.
- This soup of DNA fragments with randomly distributed lengths is then put through a filtering step called "size selection". Only DNA fragments of a certain length will remain (typically somewhere between 200-500 bp, where bp stands for base pair).
- Among many other important steps to prepare the DNA library for sequencing is "adapter ligation". This process involves tacking on special sequences to both ends of the DNA fragment which is required by the sequencing process.
- Optionally, a chemical amplification step, known as Polymerase Chain Reaction (PCR), can be conducted to increase the amount of genomic material available for sequencing.
- The sample is loaded onto a flowcell and put into an Illumina sequencer, which is capable of reading millions of DNA fragments and capturing the results digitally.
- Once the sequencing is complete, the sequencer closes the sequencing session by creating a directory of BCL files (Illumina's proprietary storage format). These are immediately converted to a FASTQ file (standard DNA storage format), typically the "input" to a computational team.
The figure below depicts how this process generates fragments of varying lengths and shows how each read is generated based on the fragments.
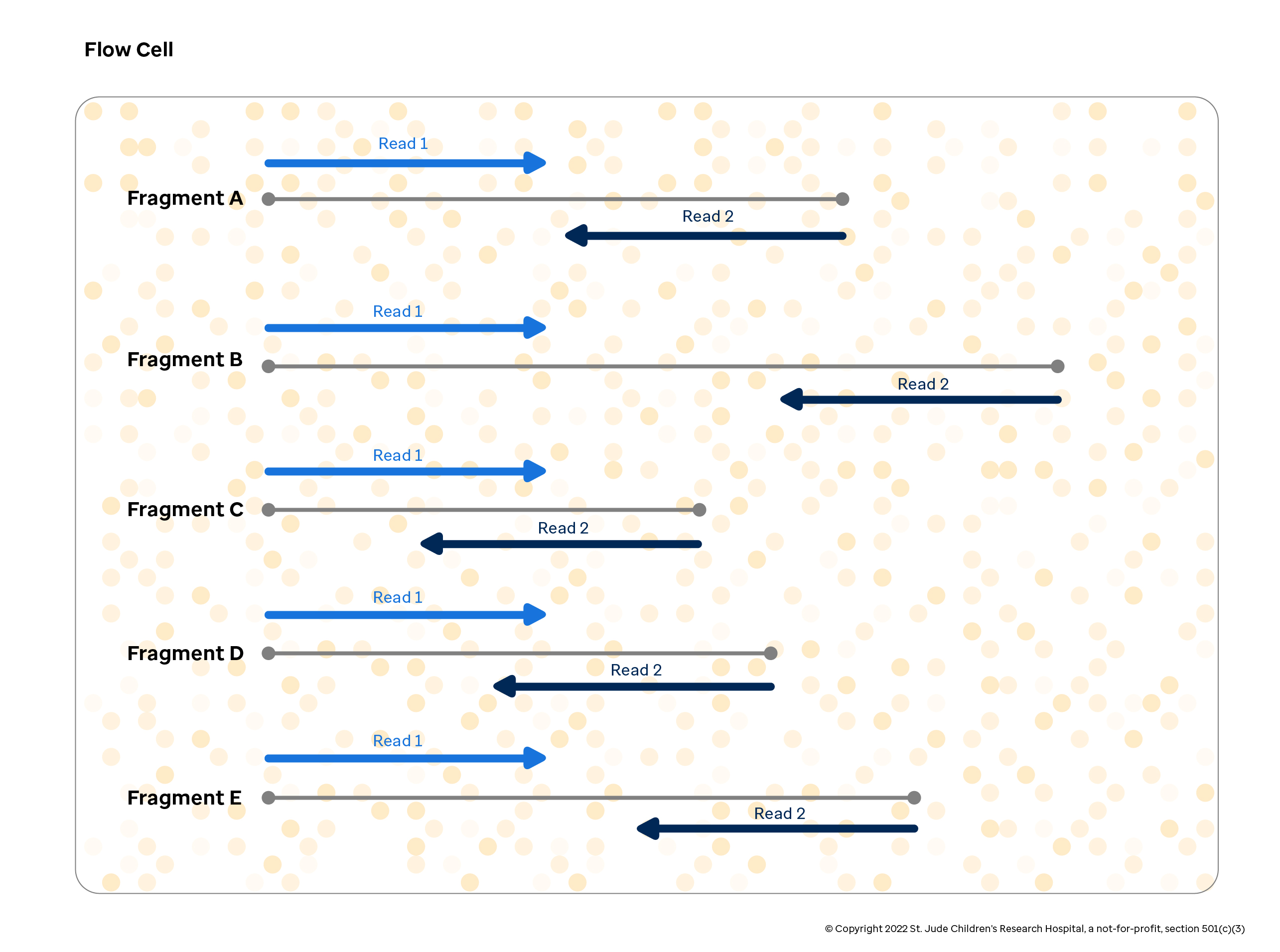
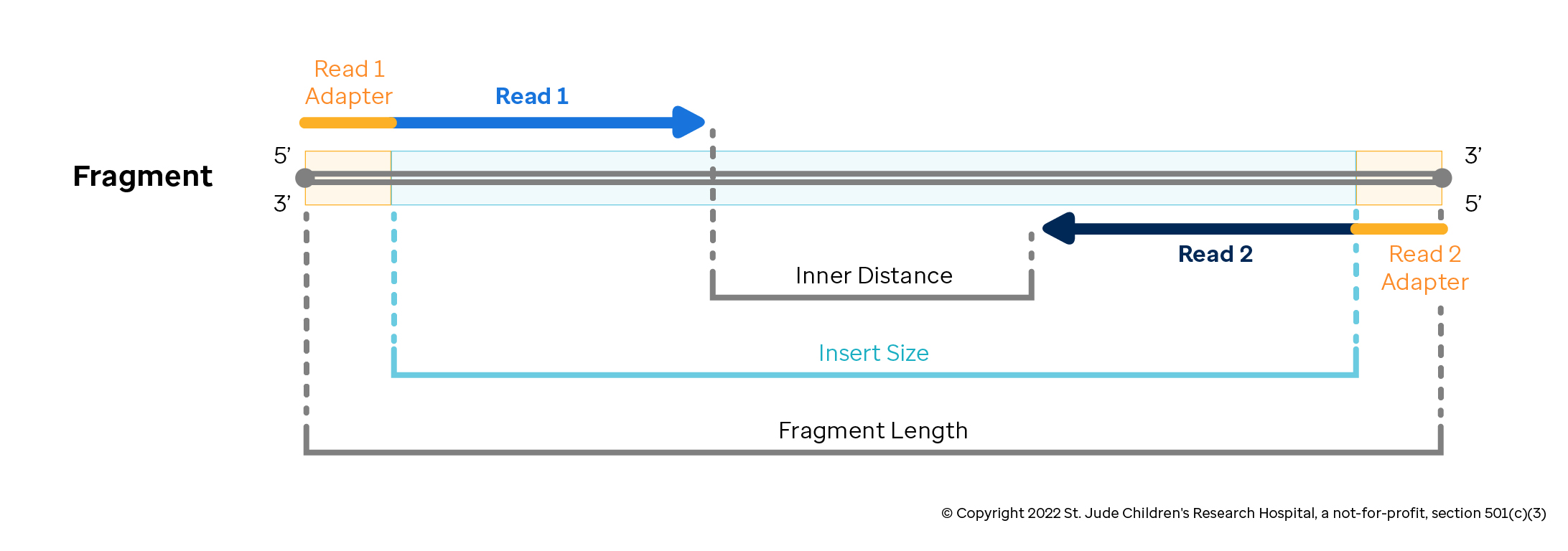
For our paired-end sequencing example, it is important to note how the DNA sequencer reads the data in step 5. In each 200-500bp DNA fragment, the sequencer begins reading from one of the fragments and drops off somewhere in the middle. It stores all of the nucleotides read from this end of the fragment with their associated confidence scores as "Read 1". After this is complete, the sequencer reads from the opposite end of the fragment and stores the nucleotide calls and confidence scores known as "Read 2". Together, the combination of a Read 1 from a DNA fragment and a Read 2 from the other end of the DNA fragment is called a "read pair" as denoted in the figure below.
After the steps above have completed, the result is a single FASTQ file (single-end sequencing) or a pair of FASTQ files (paired-end). For more information on the FASTQ file format, see its section in the "Genomic File Formats" chapter.