FASTQ Files
For most workflows in computational genomics, the input files are in the FASTQ format. The dedicated Wikipedia page for the FASTQ file is quite good, so we recommend you take a look before starting this section. As a reminder, this guide focuses on Illumina-produced FASTQ files, though the concepts are generally applicable to most sequencers today.
Technically speaking, FASTQ files are not a direct product of sequencers as many have their own intermediate representation of the reads (e.g. BCL files for Illumina sequencers) that need to be translated into FASTQ files (using a tool like bcl2fastq). In practice, many sequencing cores do this translation step before sending the data to the computational engineers downstream.
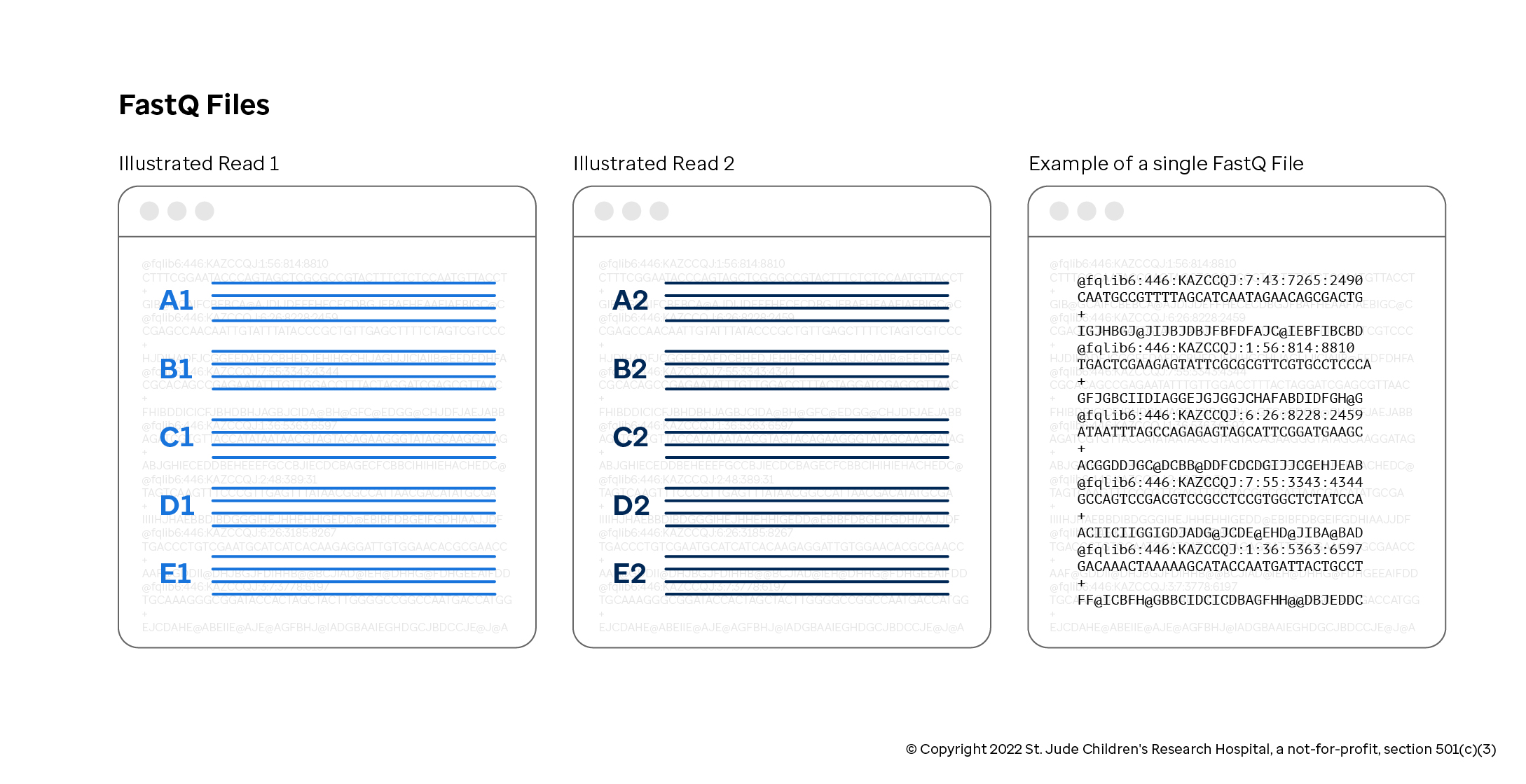
A FASTQ file may come as either a single file or an inseparable pair of files for single-end or paired-end sequencing respectively. A FASTQ file contains many reads, which, as noted before, are fragments of DNA/RNA read by the sequencer. Each read in the file has the following four-line structure separated by newlines.
| Line Number | Line Description |
|---|---|
| 1 | The sequence name, which on modern Illumina sequencers, this is a coordinate system dictating where the read originated from on the flow cell |
| 2 | The sequence of the DNA fragment |
| 3 | A single + delimiter character, often referred to as the "plus line" |
| 4 | The associated quality scores, one per nucleotide coordinating with line 2; generally an ASCII encoded probability score of incorrectness |
Random access within FASTQ files is not typical—generally, FASTQs are used
solely as input to some alignment process, which then produces a BAM file—so
they are gzipped (not bgzipped) to save space. Conventionally, FASTQ file names
indicate which read the file contains (e.g. Sample.fastq.gz for single-end
sequencing or Sample_R1.fastq.gz and Sample_R2.fastq.gz in paired-end sequencing
where _R1 stands for "read one(s)" and _R2 stands for "read two(s)").
Note that in the case of paired-end sequencing, it is crucial that each of the FASTQ files list their reads in same order. If even one read is deleted from either file, the entire read pairing will be off, which will have disastrous results during the alignment phase. To catch common formatting errors in single-end or paired-end FASTQ files, consider using fqlib (specifically, the lint subcommand).
Below is an example of the first four lines of a pair of FASTQ files generated by fq generate and validated by fq lint.
fq generate --record-count 2 Sample_R1.fastq.gz Sample_R2.fastq.gz
gzip -dc Sample_R1.fastq.gz
# @fqlib5:334:VVDJXFE:4:5:7053:1012/1
# CCTTTTCCCGCAGTCGTCAGCAGTAAGTGTGCGACCGGTAGTTCAAAAGGGGAATATCACCCGCTATTTTGCGAATACTAGAGCCTCGGTTCACGCAAGCA
# +
# GFGDDB@CJCHDBDBDB@JACCIE@J@GD@FHFADAIEA@@ABBEADHBFH@CIEFBBHGCHEJHEAIHAAIHBGEH@EDGAEEC@FGEABAHE@FAI@IC
# @fqlib5:334:VVDJXFE:7:33:2310:7985/1
# ACGTGGCCGTCCTTTTGCCAGATATCGGTAAGAGAGTTCTAGCTAAGATAATATCAATCCGCGAATGTCAGAGGGAGTGTTTCCCTTCCGGGGAAGCAAAT
# +
# GDGE@DBJJCBHCGFEBHDGEI@JA@GHEHA@CCJGCGGDJGBACIBGECDDBGBHI@GBAEBBJD@@BAFBHAHEHJCBGFGCA@GJ@IAAIJDHG@IDC
gzip -dc Sample_R2.fastq.gz
# @fqlib5:334:VVDJXFE:4:5:7053:1012/2
# ATGCCAATGGGAAGCTCGGGTAGTTCTTTCATCATGGTAATAAATCGACCGAACAACCGTTCAGGGCCAGATGAAAAGGGCCTGGCGTCTGGCACAGACCC
# +
# FGDFEHBCJFDFCFHEHFGEEADCBEF@JJD@HEGE@@AIIIDHGHADEAHA@D@DCAGBFH@HBDGICI@ICAAGJBCDDGEFJ@JBHGJFACEECJEJJ
# @fqlib5:334:VVDJXFE:7:33:2310:7985/2
# GCCCGTAGCAATACTCCTTGCGGGGACGATTATGGCGTTAATCTGATATCTCAAGCAGTAGTGGGGCTATACACTTGCGCCGTAGTCGAGCGGTTTGTAGT
# +
# DIGCJJHDCEDDDAGH@FFGHFEAB@JDA@@HH@AJJIHJCA@JFHGAHA@CCGJDF@BJ@AH@B@BH@JCCEIDCIFCED@E@D@II@HBJIADG@EFDB
fq lint Sample_R1.fastq.gz Sample_R2.fastq.gz
# Exit code is 0, which means all went well.
# Simulate a missing quality score, which would be a malformed file.
zcat Sample_R2.fastq.gz | head -n 7 | gzip -c > Sample_R2.bad.fastq.gz
fq lint Sample_R1.fastq.gz Sample_R2.bad.fastq.gz
# Sample_R2.bad.fastq.gz:8:1: [S004] CompleteValidator: Incomplete record: quality is empty