Compression and BGZF
Overview
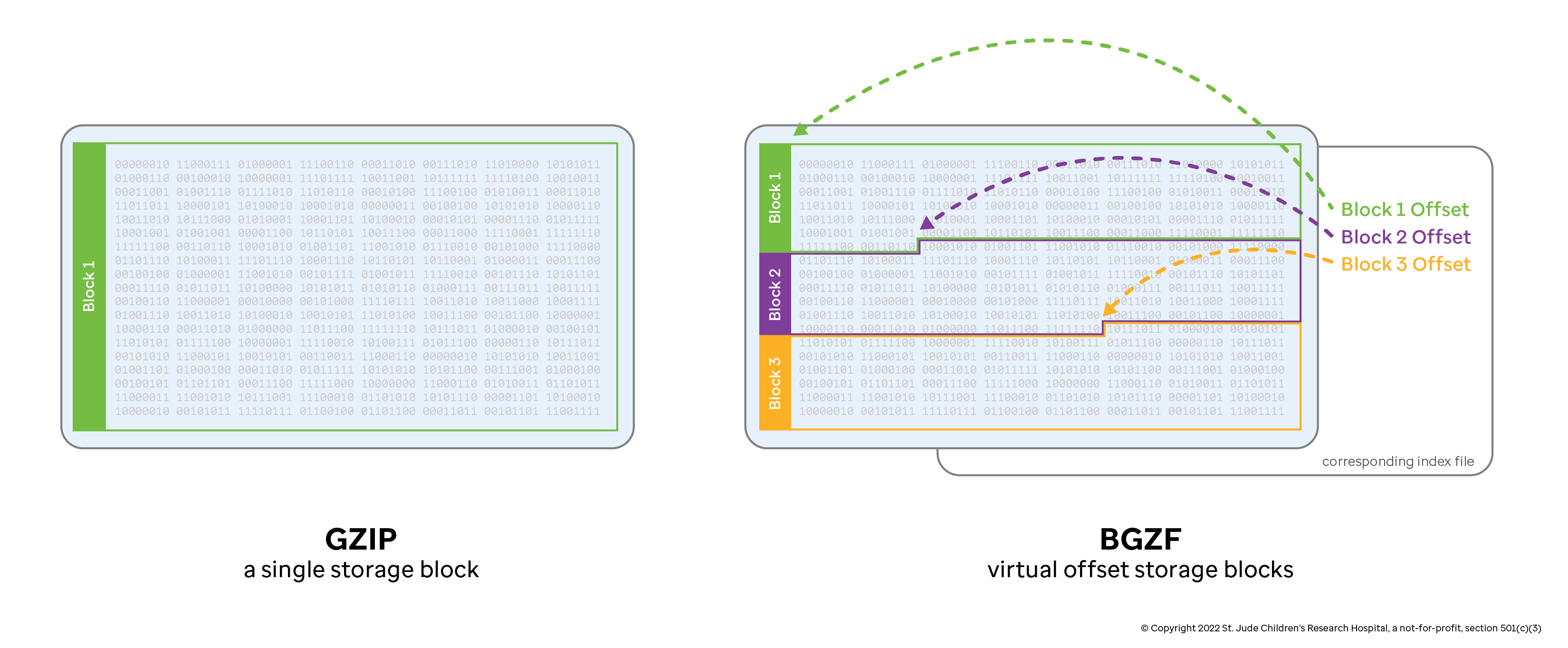
The BGZF compression technique and associated bgzip/tabix tools were developed as general purpose utilities to enable random access lookup in compressed tab-delimited text files. By default, the gzip compression algorithm creates a single stream of data—accessing information in the middle of that compressed stream requires decompressing the stream from the beginning. Genomic files tend to be substantial in size, so decompressing an entire file to retrieve information is a waste of time and resources. The BGZF compression technique solves this problem by creating multiple gzip streams as contiguous "blocks" and storing the offsets to each block in an associated index file. In this case, you can retrieve a subset of information from the file by seeking past all of the irrelevant BGZF blocks (avoiding the associated computational overhead) and only decompressing the block that contains the information you want.
Consider a single, bgzipped BAM file with the name Sample.bam. This file will
typically be accompanied by an index called Sample.bam.bai, which contains
offsets to the BGZF blocks. These two files are often treated as an inseperable
pair: without the index file, it is impossible to navigate the large BAM file
effectively (though it is trivial to generate another index, as we will see).
For detailed information on BGZF, see "The BGZF compression format" section in the SAM file specification.
Comparison with Gzip
Below is a simple comparison of gzip versus bgzip to illustrate the
trade-off. Note that while gzip achieves a slight edge in compression ratio,
the bgzip/tabix pair is vastly superior in lookup time. This non-linear
trade-off between compression and random-access speed is why BGZF is largely
pervasive in the field of computational genomics.
Worked Example
First, we'll start by downloading the GENCODE gene model v32. We'll remove the header line and then sort by chromosome name then genomic start location (numerically), as that is what
bgzipexpects.GENCODE_GTF="ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.chr_patch_hapl_scaff.annotation.gtf.gz"
curl "$GENCODE_GTF" \ # Download GTF
| gunzip \ # Decompress
| grep -v "^#" \ # Remove header lines
| sort -k1,1 -k4,4n > gencode.v32.all.gtf # Sort by chromosome name then genomic start location (numerically)Next, we'll compress the file using both
gzipandbgzip. To keep things fair but running at a decent clip, we'll choose a compression value of5for both of the commands.gzip -c -5 gencode.v32.all.gtf > gencode.v32.all.gtf.gzipped
bgzip -c -l 5 gencode.v32.all.gtf > gencode.v32.all.gtf.bgzipped
ls -lah
# Permissions Size Date Modified Name
# .rw-r--r-- 1.4G 18 Aug 12:51 gencode.v32.all.gtf
# .rw-r--r-- 64M 18 Aug 12:52 gencode.v32.all.gtf.bgzipped
# .rw-r--r-- 52M 18 Aug 12:52 gencode.v32.all.gtf.gzippedNow that we have our two files, we'll need to generate the index for the bgzip file. Here, we show the effect of trying to generate an index for the regular
gzip-ed file as well.tabix -p gff gencode.v32.all.gtf.gzipped
# [tabix] the compression of 'gencode.v32.all.gtf.gzipped' is not BGZF
tabix -p gff gencode.v32.all.gtf.bgzippedLast, let's assume we are wanting all entries for the
NOTCH1gene. Here is how you might do that in both zipping styles with the associated lookup times for each.time (zgrep "gene_name=NOTCH1" gencode.v32.all.gtf.gzipped &> /dev/null)
# ( zgrep "gene_name=NOTCH1" gencode.v32.all.gtf.gzipped &> /dev/null; ) 20.60s user 0.05s system 99% cpu 20.693 total
time (tabix gencode.v32.all.gtf.bgzipped chr9:136,496,070-136,545,786 &> /dev/null)
# ( tabix gencode.v32.all.gtf.bgzipped chr9:136,496,070-136,545,786 &> /dev/nul) 0.01s user 0.00s system 83% cpu 0.018 total